 |
 |
||||||||||
| EXAFS | ||||||||||||||||||||||||||||||||||||||||||||||||||
Abstract
During the later stages of hard-rock mining in Cornwall, UK, widespread processing and refining of arsenic in purpose-built calciners resulted in severe, localized contamination of soils with arsenic. In order to characterize the chemical speciation of arsenic in a soil depth profile of a calciner residue dump, an interdisciplinary approach was chosen. Several overlapping and complementary physical-chemical techniques were applied: X-ray powder diffraction (XRD) for detection of crystalline phases, sequential extraction combined with hyphenated speciation methods for measurement of the association of arsenic species with soil mineral phases, and X-ray absorption spectroscopic (XAS) methods such as XANES (x-ray absorption near edge structure) and EXAFS (extended x-ray absorption fine structure) for revealing arsenic molecular information, i.e. oxidation state and local structure around As atoms.
Total arsenic concentrations ranged from 0.29 to 15.4 % m/m, with higher concentrations in the deeper layers. Arsenic was predominantly present in pentavalent form, bound to amorphous or poorely-crystalline hydrous oxides of Fe (probably ![]() -hematite). In the soil samples with the highest arsenic concentrations, a small amount of a non-classified crystalline iron arsenate phase was found, viz.
-hematite). In the soil samples with the highest arsenic concentrations, a small amount of a non-classified crystalline iron arsenate phase was found, viz.
Fe![]() (As(AsO
(As(AsO![]() )
)![]() ), which contains both tri- and pentavalent arsenic in the crystal structure. There was also evidence for the presence of some arsenate bound to quartz (
), which contains both tri- and pentavalent arsenic in the crystal structure. There was also evidence for the presence of some arsenate bound to quartz (![]() -SiO
-SiO![]() ). In the deepest layers, the coordination of arsenic to Fe and Si atoms was much more pronounced than in the highest layer, yielding them much less mobile. The overall results make us believe that the normally assumed relative safety, from a mobility point of view, is questionable since only a relative small fraction of arsenic is found in a crystalline iron arsenate form.
). In the deepest layers, the coordination of arsenic to Fe and Si atoms was much more pronounced than in the highest layer, yielding them much less mobile. The overall results make us believe that the normally assumed relative safety, from a mobility point of view, is questionable since only a relative small fraction of arsenic is found in a crystalline iron arsenate form.
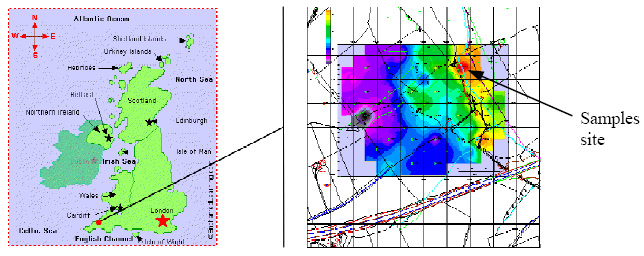
Figure 1: Samples were taken near a calciner in Cornwall, located ca. 4 kmwest of the Camborne-Redruth-St. Day orefield, which ceased operation in the 1920s. The spatial distribution of total arsenic values in the soil areindicated by the color scale. The exact sampling location is the calciner's residue dump as indicated by the arrow, with alarmingly high soil arsenic concentrations of more than 10%.
From prehistorical times until recent years, polymetallic ores in Cornwall (UK) were commercially mined. Although the main products were copper and tin, many other metals and metalloids were also recovered. Initially, arsenic was vented to the atmosphere as a deleterious element during tin ore smelting. This led to the dispersion of arsenic over an area of approximately 700 km![]() , with arsenic levels in agricultural and garden soils as high as 1000 mg kg
, with arsenic levels in agricultural and garden soils as high as 1000 mg kg![]() and sometime more (Abrahams and Thornton, 1987). When applications for arsenic, including pigments, pesticides, and as a glass decoloriser, were discovered in the early 1800s, the metalloid was recovered and refined in purpose-built calciners. Nevertheless, the arsenic production was phased out rapidly at the beginning of the twentieth century. The primary arsenic-bearing ores used for arsenic production were arsenopyrite (FeAsS) and löllingite (FeAs
and sometime more (Abrahams and Thornton, 1987). When applications for arsenic, including pigments, pesticides, and as a glass decoloriser, were discovered in the early 1800s, the metalloid was recovered and refined in purpose-built calciners. Nevertheless, the arsenic production was phased out rapidly at the beginning of the twentieth century. The primary arsenic-bearing ores used for arsenic production were arsenopyrite (FeAsS) and löllingite (FeAs![]() ), which were roasted in the calciner where arsenic sublimated and condensated as arsenolite (As
), which were roasted in the calciner where arsenic sublimated and condensated as arsenolite (As![]() O
O![]() ) in collection chambers. The subsequent dumping of residues created hot spots with alarmingly high soil arsenic concentrations in the vicinity of calciners.
) in collection chambers. The subsequent dumping of residues created hot spots with alarmingly high soil arsenic concentrations in the vicinity of calciners.

Figure 2: Site overview: Camborne-Redruth-St. Day mining district.
In spite of such hot spots and generally elevated arsenic levels in soil, neither drinking water (DWI, 2002) nor locally grown crops (Warren et al., 2003) appear to pose a real danger to the public. It should be noted that arsenic levels measured in urine of Cornwall residents were found to be higher than from a non-Cornish control group (Kavanagh et al., 1997). On sites historically contaminated with arsenic, weathering of primary arsenic minerals has been reported to lead to the formation of secondary, more thermodynamically stable, arsenic minerals such as e.g. hornesite (Mg![]() (AsO
(AsO![]() )
)![]() .8H
.8H![]() O) (Voigt et al., 1996), scorodite (FeAsO
O) (Voigt et al., 1996), scorodite (FeAsO![]() .2H
.2H![]() O) (Foster et al., 1998), etc.. Recent work (Matera et al., 2003) has shown that arsenic in soil from former industrial sites may be potentially mobile because of its association to amorphous iron oxides that could be solubilized in reducing conditions. The fixation of arsenic in these soils was hypothesized to be either by precipitation, corresponding to formation of secondary minerals/phases such as iron arsenates, or by sorption of arsenate (As(V)) on iron (hydr)oxides with an arsenic oxyanion immobilization by iron (hydr)oxides. The mobilization risk is more pronounced for arsenite (As(III)) than for arsenate since arsenate binds more strongly to minerals (Pierce and Moore, 1982). In some instances the leaching of other arsenic compounds such as monomethylarsonic acid (MMAA) and dimethylarsinic acid (DMAA) has been reported (Montperrus et al., 2002). It is clear that management of such highly polluted sites is mandatory in the interest of public health.
O) (Foster et al., 1998), etc.. Recent work (Matera et al., 2003) has shown that arsenic in soil from former industrial sites may be potentially mobile because of its association to amorphous iron oxides that could be solubilized in reducing conditions. The fixation of arsenic in these soils was hypothesized to be either by precipitation, corresponding to formation of secondary minerals/phases such as iron arsenates, or by sorption of arsenate (As(V)) on iron (hydr)oxides with an arsenic oxyanion immobilization by iron (hydr)oxides. The mobilization risk is more pronounced for arsenite (As(III)) than for arsenate since arsenate binds more strongly to minerals (Pierce and Moore, 1982). In some instances the leaching of other arsenic compounds such as monomethylarsonic acid (MMAA) and dimethylarsinic acid (DMAA) has been reported (Montperrus et al., 2002). It is clear that management of such highly polluted sites is mandatory in the interest of public health.

Figure 3: Remains of the mines in the Camborne area.
The potential leaching behaviour of arsenic is usually studied by sequential extraction protocols which give insight into the association with soil phases. However, such an approach only yields very crude information on the arsenic binding and gives no structural information on the As-atoms surroundings, which is crucial to assess the potential toxicity and mobility risks [3,4]. To this end, we applied x-ray absorption spectroscopic methods XANES (x-ray absorption near edge structure) and EXAFS (extended x-ray absorption fine structure) to retrieve arsenic molecular information, i.e. oxidation state and local structure around As atoms in the soil. X-ray absorption spectroscopy is of particular significance for the characterization of arsenic on poorly-crystallized materials because most other structural probes either require long-range ordered materials, or cannot probe the precise vicinity of the adsorbate (Matera et al., 2003) In this way the most abundant modes of As bonding can be identified [2-10], among which different crystalline and amorphous solid phases as well as surface-bound or adsorbed As species on mineral particles may coexist, as shown in previous analyses of industrially polluted soils [2-8].

Figure 4: Section through a mine in the 1770s.
In this study, the residue dump near a calciner was selected for characterization of arsenic in the soil. Samples from this location were previously studied by SEM-EDS and were found to contain ca. 12% arsenic, mostly present as As-Fe oxides (Camm et al., 2003). For characterization, we have chosen a series of multifold (multidimensional) analytical approaches which both overlap and complement each other in order to obtain as much arsenic speciation information as possible. Samples from a soil depth profile and a mixed soil sample from the aforementioned location were analysed using X-ray powder diffraction and X-ray absorption spectroscopy combined with sequential extraction and hyphenated speciation techniques.

Figure 5: Typical mine scene in Cornwall at the beginning of 20th century.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
E-mail:iztok.arcon@p-ng.si Last change: 28-Jun-2006 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||